CHAPTER 3 - CELL SIGNALING INITIATED FROM CD40
3.1 CD40 clustering and downstream signaling
3.1.1 Consequences of membrane receptor crosslinking in cell signaling
As shown in chapter 1, CD40 crosslinking on B cell is a prerequisite for their activation. Anti-CD40 mAb F(ab) fragments are less active, and non-crosslinked anti-CD40 mAb can induce B cell proliferation only in the presence of a costimulus. Moreover, soluble CD40L in trimeric form activates B cell potently in the presence of costimuli, whereas membrane form is directly mitogenic. These results confirmed preceding observations showing requirement of CD40 aggregation for optimal signaling.[1] Importantly, CD40L clustering on T cells, upon initial contact with its cognate receptor, is required for aggregation of CD40 on B lymphocytes.[2]
Bjorck et al. proposed that both CD40 crosslink degree and efficiency might determine strength of the cell signaling.[3] This crosslinking requirement has also been observed with other members of the TNF-R family. For example, Fas signaling requires hexameric FasL for organization of the death−inducing signaling complex (DISC), in which receptor oligomerization increases its avidity for FasL, and the efficient induction of apoptosis.[4]
3.1.2 Minimal CD40 oligomerization requirement for activation of cell signaling
3.1.2.1 Extracellular oligomerization
Although monomeric CD40L is unable to induce B cell proliferation, it binds to CD40, upregulates expression of class II molecules,[5] and promotes rescue of B cells from apoptosis.[6] This suggests that the receptor crosslinking level necessary for the initiation of some responses might vary among type and differentiation stage of a cell.[7] This is consistent with observation that, at levels higher than 5 × 105 of CD40 molecules in a transfected cell, the NF-κB pathway is activated in the absence of CD40L.[8]
Moreover, hexameric and dodecameric forms of CD40L were shown to be more potent than the trimeric form in induction of B cell proliferation and expression of costimulatory molecules,[4],[9] although these two forms induced similar level of MHC II molecule expression. In contrast, trimeric and hexameric CD40L were shown to activate similar levels of NF-κB and p38 pathways in CD40−transfected cells.[10] Surprisingly, aggregated anti-CD40 mAb as well as soluble trimeric CD40L were unable to mediate IL-6 production in B cells, in contrast to membrane-bound CD40L.[11] Thus, insufficient CD40 crosslinking could not explain entirely the non-activity of anti-CD40 mAb.
In the case of CD95, apoptosis is induced by agonistic mAb as efficiently as by natural ligand. Since CD95 microaggregation is induced by agonistic mAb but not by CD95L, receptor clustering is not a prerequisite for initiation of cell death.[12] This suggests that CD40 aggregation could not account alone for the differences in cell signaling activated by diverse stimuli.
3.1.2.2 Intracellular oligomerization
During transfection experiments, dimeric construct of the CD40 cytoplasmic domain was shown to be sufficient for activation of the NF-κB pathway, although trimerization generated a stronger signal.[13] Targeting of trimeric domain to the membrane by meristoylation provided maximal activation of NF-κB. Accordingly, disulfide-linked CD40/CD40 homodimers[14] were observed in CD40−transfected and EBV−positive cells, and their levels rapidly increased upon CD40 oligomerization by either trimeric CD40L or crosslinked anti-CD40 mAb.[15] Interestingly, CD40/CD40 dimer formation was demonstrated to be critical for some but not all cellular events. It could thus serve as initiator for rapid extension of CD40 oligomerization, bringing receptor molecules into close proximity with one another, what is needed for certain biological functions.
3.1.3 Structural mechanisms
governing initiation of intracellular signals
Different anti-CD40 mAbs were tested for competitive binding on B cells in the presence of soluble trimeric CD40L. Whereas all of them promoted B cell rescue from apoptosis, indicating that this phenomenon does not depend on the CD40 epitope, only those competing for the CD40L binding site induced homotypic adhesions.[6] Furthermore, mAbs that poorly inhibited CD40L binding to CD40 synergized with soluble CD40L−induced B cell proliferation in the absence of costimulus. Similarly, non-competing mAbs were shown to synergize with soluble trimeric CD40L for promoting CD23 expression and IgE production in human B cells.[16] Additional data indicated that a monomeric scFv derived from the anti-CD40 mAb G28-5 is a potent agonist, able to crosslink the receptor at the membrane, and cooperates with soluble natural CD40L.[17] Moreover, epitope specificity of these scFv has an important role in defining the functional properties of the receptor.[18] All these observations suggest the existence of some allosteric movements and cooperativity between the different epitopes, in which formation of CD40-complex induced by the binding of the first ligand may influence the binding of subsequent molecules. This might ultimately lead to an enhanced binding of CD40L and/or stabilization of the CD40–CD40L complex.
Surface plasmon resonance experiments confirmed the correlation between the ability of anti-CD40 mAbs to compete with CD40L for binding to CD40 and cellular activation. More importantly, these experiments also demonstrated the absence of relation between high affinity of the mAbs and their activation potential.[19] Thus, signaling pathways activated downstream CD40 are also determined by the manner CD40 is ligated.
Only the natural CD40–ligand, both in the membrane and soluble forms, may strictly provide the epitopes required for changes of the CD40 conformation that are necessary for the engagement of dedicated pathways. This cannot be fully imitated by the binding of anti-CD40 antibody. In contrast, Ellmark et al. showed that the extracellular domains of human CD40 are actually not essential for rescue of IgM−induced B cell line apoptosis.[20] Finally, the precise mechanisms of oligomerization and molecular activation of CD40 at the cell membrane upon its ligation remain to be elucidated.
3.2 Role of pre-ligand binding assembly domain in CD40
3.2.1 The PLAD domain
It has been observed that some members of the TNF-R family assemble constitutively in the absence of their cognate ligand. Papoff and coworkers first demonstrated that ligand–independent oligomerization of CD95 depends on the most distal extracellular domain CRD1 but not on the death domain (Figure 19A).[21] Whereas soluble form of the receptor homo–oligomerizes, and hetero–oligomerizes with the membranous form, it is not the case for a truncated form lacking the 42 N-ter residues that compose most of the CRD1. Capacity to interact with CD95L and to initiate signaling when overexpressed at the membrane was conserved by this truncated form. Finally, a soluble truncated form of CD95 made of the 49 N-ter amino-acids is necessary and sufficient to mediate oligomerization of the receptor.
Similarly, TNF-R1 and R2 self–associate in the absence of TNFα, as seen by fluorescence resonance energy transfer on living cells.[22] The putative N-ter functional domain that mediates ligand-independent receptor association was named pre–ligand binding assembly domain (PLAD). The PLAD is responsible for preformed receptor oligomerization prior to ligand binding, what is not sufficient for initiation of intracellular signals, but is necessary for the interaction with the cognate ligand (Figure 19A,B). Recently, TNF-R PLAD was targeted in the treatment of inflammation.[23] A soluble construct of TNF-R1 PLAD was demonstrated to bind to the receptor (Figure 19C), to potently inhibit effects of TNFα and consequently, to ameliorate inflammatory arthritis in various experimental mouse models by blocking TNFα–induced NF-κB activation, expression of RANK and RANKL, and osteoclast differentiation.
These observations give explanations for the resistance of lymphocytes from patients with autoimmune lymphoproliferative syndrome to Fas-induced apoptosis.[24] Receptor molecules mutated in either the ligand–binding domain or in the death domain, still oligomerize with wild–type receptor, forming mixed trimers. These defective molecules thus dominantly interfere with apoptosis induced by CD95L, but not with apoptosis mediated by agonistic anti-CD95 mAb.
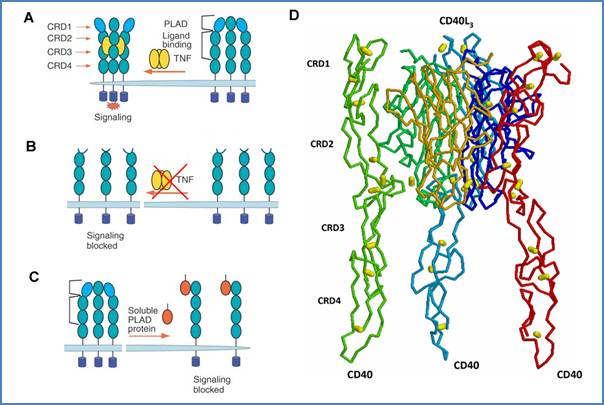
Figure 19. Implication of PLAD in TNF-R and CD40 signaling. PLAD is necessary for pre-formed trimer upon ligation (A). Mutated PLAD prevents binding of TNF to the receptor (B). Soluble PLAD protein dominantly interferes with TNF-R signaling (C). Figures 19A,B,C are adapted from Deng et al.[23] CD40 PLAD may be located in CRD1 as seen on the CD40–CD40L interaction model (D). CD40 backbone chains are colored in red, light green and light blue, cysteines are represented by yellow balls.
3.2.2 PLAD requirement for CD40 signaling
Although the complete mechanism of CD40–induced signaling is not fully understood, the simplest ligand-induced oligomerization model is not sufficient to explain initiation of downstream pathways. Increase in the avidity of the receptor–ligand interaction and/or change in the membrane arrangement of the receptor that allow binding of adaptor molecules and activation upon ligation of the receptor, are certainly implicated.
CD40, as well as TRAIL-R1 (DR4), were also shown to specifically pre–associate at the membrane in the absence of ligand.[23] This confirmed earlier observations of the CD40 oligomeric state in unstimulated cells.[14] Malmborg Hager and Ellmark have further studied the ligand-independent oligomerization of CD40.[19] They showed that CD40L binding to its receptor requires the CRD1 of CD40, where PLAD might be located based on homology studies (chapter 2), although this domain does not contain any of the residues important for CD40L ligation (Figure 19D).
Moreover, CD40 signaling was shown to be independent of the CRD1, since ligation of engineered CD40 proteins lacking part of their extracellular domains, by a peptide tag is sufficient to rescue anti-IgM–induced apoptosis of WEHI cells.[20]
3.3 CD40 signaling is initiated within membrane rafts
3.3.1 Membrane rafts
The outer leaf plasma membrane of many cell types contains microdomains of approximately 50 nm of diameter, named lipid or membrane rafts. These microdomains structures move within the fluid bilayer and constitute functional platforms for the formation of signalosomes.[25] They are enriched in sphingolipids and cholesterol that self-aggregate and segregate from bulk unsaturated glycerophospholipids, forming a thick liquid-ordered phase. This resists to solubilization in non-ionic detergent, but can be isolated from low density fractions after buoyancy in a discontinuous sucrose gradient. Similar cholesterol–rich rafts exist in the inner leaflet of the plasma membrane, but their composition and function is not yet well known.[26]
These microdomains have been described as essential structural platforms for a variety of functions. For example, lipid rafts have been shown to be crucial for T cell apoptosis mediated by Fas following TCR stimulation.[27] Modulation of TNF-R family protein location into lipid rafts may dynamically regulate the efficiency and outcomes of signaling by these receptors.
3.3.2 CD40 is localized into lipid raft microdomains
Ligand–independent pre-associated CD95 complexes were preferentially found within lipid rafts.[27] Similarly, Hostager et al. have shown that a significant level of CD40 is constitutively localized in these lipid microdomains in unstimulated mouse B cell line.[28] Similarly, CD40 engagement induces the nearly complete translocation of CD40 from detergent−soluble to detergent−insoluble fractions of plasma membrane, and this independently of downstream signaling molecules. Same observations have been reported on human DCs, in which integrity and reorganization of membranes rafts is required for engagement of signaling from CD40.[29] Although Malapati and co-workers described that CD40 functions outside the lipid rafts during BCR signaling in murine B lymphoma,[30] most of the authors gave consistent data showing that co-engagement of BCR and CD40 brings their signaling complexes into close proximity within rafts microdomains, allowing crosstalk between them.[31] CD40L colocalized with CD40 within membrane rafts in unstimulated non-Hodgkin’s lymphoma B cells,[32] what was thought to be responsible for the constitutive activation of NF-κB and autonomous cell growth in B cell lymphomas (chapter 2).
Targeting of CD40 C-ter signaling domain to lipid rafts and artificial trimerization were shown to synergistically initiate cell signaling, suggesting that both localization within lipid rafts and trimerization of the receptor are important for activation of CD40-induced signaling.[8] Finally, one can suggest that binding of CD40L may induce a conformational change in the CD40 transmembrane domain, allowing it to interact with ceramide of the membrane rafts,[33] linking allosteric movements engaged by CD40L to location within lipid rafts.
3.3.3 Caveolae are implicated in CD40 signaling
Caveolae are lipid rafts structures coated with caveolin that form invaginations of the plasma membrane. In contrast to coated-pits, these domains are not constitutively internalized.[25] CD40 was detected in caveolae together with some key signaling intermediates in unstimulated human renal proximal tubule cells.[34] CD40 engagement induces its dissociation as well as the dissociation of signaling partners from this structure, what is necessary for induction of downstream pathways.
3.3.4 Translocation of CD40 into lipid rafts
CD40 clustering was shown to be dependent on acid sphingomyelinase (ASM) translocation from intracellular stores onto the outer leaflet of the cell membrane,[35] in the same manner as CD95.[36] Primary stimulation via CD40 induced activation and membrane translocation of ASM which then colocalizes with the receptor and mediates release of ceramide. ASM accumulates in preexisting sphingolipid−rich rafts and triggers the formation of larger ceramide-enriched platforms, bringing together receptor and signaling molecules into close contact. Moreover, CD40–clustering causes stabilization of the ligand–receptor interaction leading to sustained signaling (Figure 20).[37] Mechanisms of ASM activation are still poorly understood. Caspases have been proposed to mediate upstream events initiating by CD95.[38] Recently, PKCδ was shown to activate ASM.[39]
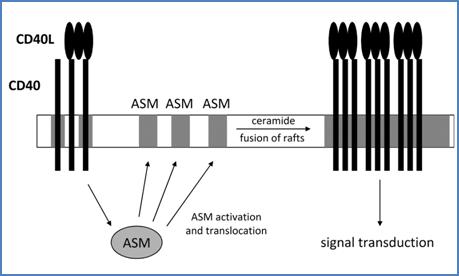
Figure 20. CD40 reorganization into rafts is dependent on ASM. Primary CD40 triggering with its cognate ligand induces activation and translocation of ASM to the plasma membrane (left). ASM thus induces fusion of rafts microdomains and oligomerization of CD40 molecules. This step is essential for CD40L−induced cell signal transduction. This scheme can be viewed with CD40 and CD40L inverted (see text below).
In addition to primary activation of ASM from CD40, pre–clustering of CD40L is required for CD40 aggregation.[2] As for CD40 (Figure 20), CD40L ligation was shown to induce ASM activation and translocation to the membrane, leading to clustering of CD40L in ceramide−rich rafts.
3.4 CD40−induced cell signaling
3.4.1 CD40–activated signaling
pathways
3.4.1.1 Intermediates implicated in CD40 signaling
Although CD40 phosphorylation was observed in B cells,[40] no receptor–autophosphorylation activity was detectable.[1] CD40 phosphorylation, that was enhanced in PMA–activated human tonsil B cells, involves serine and/or threonine since no tyrosine residue is present in the cytoplasmic domain (Chapter 2, Figure 1).[41]
Triggering of CD40 was shown to induce rapid serine and threonine phosphorylation on various proteins.[42] It was also linked to protein tyrosine kinase/protein phosphatase activities.[43] In contrast to signals transduced through sIg,[44] CD40–induced tyrosine phosphorylation does not depend on activation of PKC, or on calmodulin– and calcineurin–dependent kinases,[45] but is important for stimulation of phosphoinositide (PI) turnover. Cell activation state also greatly influences CD40–induced events downstream the receptor, as seen for example in germinal center B cells where CD40 is coupled to functional protein tyrosine kinase but not the phosphatidylinositol signaling pathway.[46]
3.4.1.2 Common signaling pathways activated by CD40
Protein tyrosine phosphatases are also implicated in the early events of CD40 signaling in human B cells, and are for example involved in the regulation of protein tyrosine kinases Lyn, Fyn and Syk,[47] as well as others PTKs.[45] Janus kinase 3 (JAK3), phosphatidyl inositol-3 kinase (PI3K), phospholipase Cγ2 (PLCγ2) are also implicated in CD40 signaling. Second messengers like IP3, calcium as well as cyclic adenosine monophosphate (cAMP) have also been detected following CD40 activation.
T cell−dependent B cell stimulation is associated with NF-κB induction via the CD40–CD40L interaction, independently of PKC−mediated signaling,[48] but dependent on both protein tyrosine kinase−dependent pathways and the redox equilibrium of the cell.[49] Noteworthy, engagement of CD40 on human Daudi cells by both whole anti-CD40 and F(ab’)2 fragments led to the same level of NF-κB activation.
Numerous additional studies have described various signaling pathways engaged by CD40 that are summarized at the end of this chapter as exhaustively as possible from the current literature (Poster 1). Signaling pathways described in the most studied cell systems were included. I have paid attention on the cell–type in which pathways have been described. Although many pathways have been found in more than one cell–type, some others are cell–type–specific. Key intermediates that might contribute to CD40–signaling were also added. Due to high complexity and heterogeneity of the systems used, with sometimes controversial results, these pathways are not discussed here. The reader is invited to refer to corresponding references for detailed data since description of all these pathways is not the purpose of the thesis. Nevertheless, this figure underlines that activation of CD40 engages many common signaling pathways, and other remain certainly to be discovered. Targeting CD40 to induce one desired signal in one selective cell type is thus far from being realistic for the moment.
3.4.2 Implication of TRAF adaptor
molecules
3.4.2.1 CD40 signals through adaptor molecules
CD40 cytoplasmic domain is relatively small (Figure 1, chapter 2) and does not exhibit intrinsic enzymatic capacity. In contrast to some of the TNF-R family members, like Fas and TNF-R1 which contain an 80-residues death domain involved in caspase−mediated induction of apoptosis,[50] CD40 does not present obvious signaling motifs in the cytoplasmic domain. Nevertheless, the cytoplasmic region around Thr234 was shown to be essential in the signal transduced through CD40.[51] Thus, external CD40 stimuli are transduced upon reorganization of the receptor and other different intermediate molecules under the membrane.
3.4.2.2 The TNF-R–associated factors
3.4.2.2.1 The TRAF family
The most important intracellular signaling proteins that interact with CD40, as well as with many members of the TNF-R family, are the TNF-R–associated factors (TRAFs). These adapter proteins are constituted of 409 to 567 amino acids and have no intrinsic enzymatic activity. The TRAF family is a phylogenetically conserved group of scaffold proteins that link receptors of the TNF-R and Toll/IL-1 family to signaling cascades.[52] To date, six different members have been identified in mammalians, two in Drosophila and one in Caenorhabditis elegans (Figure 21).[53] A homologous protein designated TRAF7 was recently identified. It is involved in apoptosis, and mediates TNFα–induced activation of p38, JNK pathways and regulation of NF-κB and AP-1 via MEKK3.[54] But its implication in other TNF-R pathways has not been reported for the moment.
Interestingly, the main Epstein-Barr virus oncogenic latent membrane protein 1 (LMP1) was shown to mimic a constitutive CD40 that lacks an extracellular ligand. LMP1 interacts with TRAFs 2, 3, 5 and 6 and continuously initiates NF-κB, JNK/AP-1, p38 and PI3K pathways.[55] Although differences exist between pathways activated by CD40 and LMP1, CD40–induced cell signaling may be influence by LMP1.[56]
3.4.2.2.2 TRAFs are located into lipid rafts
Interestingly, TRAFs 2 and 3 were shown to be recruited into membrane rafts subsequently to CD40 activation.[58] Signals induced from other receptors can either positively or negatively influence TRAFs recruitment to the signalosome.[57] Duration and strength of TRAF2–dependent signaling was shown to be regulated by degradation of TRAF2 that is directed by CD40–induced ubiquitination.[58] Finally, TRAFs compartmentalization within rafts was shown to occur subsequently to their CD40–mediated recruitment at the membrane. Stabilization of the receptor signaling complex, and positioning of all signaling partners into vicinity, ultimately form the signalosome.[2]
3.4.2.2.3 Regulation of TRAF functions
Expression of TRAF members is regulated in a cell–specific manner at the transcriptional level.[55] Post-transcriptional modulation also occurs, since different spliced forms of TRAF2 were reported.[59] Finally, post–translational down–regulation of TRAF2 by CD40-mediated degradation/depletion has been observed,[28] as well as TRAF3 stabilization, what leads to its upregulation in urothelial cell CD40L–stimulated carcinoma.[60] TRAF2 also plays an important role in the activation–induced degradation of TRAF3.[61] TRAF1 was shown to redistribute TRAF2 and CD40 from raft microdomains to detergent–soluble fractions, and to protect TRAF2 from CD40–mediated degradation.[62] Relative intracellular abundance of TRAF proteins, as well as their interplay, play a critical role in modulation of biological responses induced through them.
3.4.2.2.4 TRAF protein structure
All members of the TRAF family have a conserved C-terminal domain named the TRAF domain, itself subdivided into a highly homologous C-ter region termed TRAF-C and a region termed TRAF-N (Figure 21). The TRAF-N region possesses a coiled-coil domain and is involved in the interactions of TRAF molecules with other proteins found in the TRAF-receptor complex.[63] The TRAF-C region is critical for the interaction of TRAFs with the cytoplasmic domain of their respective receptors.[64] However, the TRAF-N also contributes to these associations.
All TRAF proteins except TRAF1 have an N-terminal RING finger, and all possess one (TRAF1) or several (TRAF2-6) zinc fingers (Figure 21). These structural motifs are important in promoting downstream signaling.[60] Moreover, zinc coordination by cysteine in RING and zinc finger motifs is essential for DNA binding. Although the N-terminal half of TRAF2 has been shown to translocate into the nucleus and to transactivate gene transcription in transfected cells,[65] whether or not TRAFs directly regulate gene transcription in vivo remains uncertain. Concerning TRAF7, except for N-terminal RING and zinc finger domains, it differs from the other members in the C-ter region that contains WD40 repeats and not the TRAF domain (Figure 21).
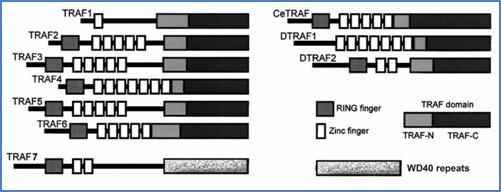
Figure 21. The TRAF family members. Schematic representation of the TRAF family proteins and their functional domains. Figure is adapted from Wajant et al.[53]
TRAFs 3 and 5 have an additional isoleucine zipper motif between the most C-terminal zinc finger and the TRAF domain that can mediate homo- and heteromultimerization of these proteins.[66] In contrast, TRAFs 1 and 2 homo– and hetero–oligomerize through the TRAF-C domain.[67] Crystal structures of TRAF2’s TRAF domain indicate that TRAF-C forms an eight−strand antiparallel β−sandwich structure and TRAF-N forms a single α−helix (Figure 24).[68] Three TRAF2 molecules spontaneously multimerize through their TRAF domains to form a mushroom like trimer, in which TRAF-N assemble around a flexible triple helical coiled-coil, and the TRAF-C form a unique edge (Figure 22).

Figure 22. Three-dimensional structure of the human TRAF2’s TRAF domain. Spacefill representation of TRAF domain of TRAF2 assembled in trimer. TRAF-C domain (residues 356-501) and part of the TRAF-N domain (residues 272-355) are shown. Residues composing β–strands are colored in yellow, and those composing α−helixes are colored in pink (PDB code 1QSC).[68]
3.4.2.3 The TRAF–CD40 interaction
3.4.2.3.1 TRAF–binding sites
TRAF–CD40 interaction is mediated by specific sites on both proteins. Earlier studies have demonstrated that TRAF6 binds to the membrane–proximal linear motif 231QEPQEINF238 of the CD40 intracytoplasmic domain (Figure 25).[69] A distinct binding site was described for TRAFs 1, 2, 3 and 5, which interact either directly or indirectly with CD40.[70] This motif was mapped on residues 250PVQET254 (Figure 25).[72] Further findings suggested that TRAF2 and TRAF3 recognize distinct residues within the PVQET motif.[71] Since both TRAF2 and TRAF3 have been immunoprecipitated with an anti-CD40 mAb in human B cells, the simultaneous recruitment of both TRAF proteins to CD40 is possible.[60] Recently, a second functional TRAF2 binding site was identified at the C-terminal end of CD40, that consists on the 272SVQE275 motif (Figure 25).[72]
Nevertheless, these TRAF binding sites have distinct contributions. Indeed, in primary DCs TRAFs 2, 3, 5 and 6 all contribute to DC maturation, but only TRAF6 is essential for activation of p38 and JNK pathways and production of IL-12 p40.[73] Furthermore, different TRAF proteins might be implicated in distinct signaling pathways at different stages of the B cell development, as suggested for TRAF6 which plays a crucial role in affinity maturation and long-lived plasma cell,[74] whereas TRAF2 and 3 are essential for class switching.[75]
Interestingly, overexpression of the I-TRAF/TANK protein, which contains a PXQXT motif,[76] inhibited NF-κB activation induced by overexpression of TRAF2. In contrast, its expression in small amounts enhanced TRAF2–induced NF-κB signaling, suggesting that I-TRAF/TANK might not only act as an inhibitor of TRAFs, but also serve as a scaffolding adaptor linking TRAFs with important downstream partners.[77]
3.4.2.3.2 CD40–mediated activation of TRAFs
TRAFs 1, 2, 3 and 5 have similar specificity of binding to TNF-Rs.[78] Moreover, a unique motif derived from the cytoplasmic domain of CD40 was shown to bind to both TRAF2 and TRAF3. These observations have led to the questions of specificity and modulation of each TRAF function during CD40 signaling. CD40 cytoplasmic fusion protein binds to TRAF2 and TRAF3 with a low affinity, as measured by surface plasmon resonance.[10] No binding was detected to TRAF1 and TRAF6. Importantly, TRAF trimerization was required for high–affinity interaction with CD40, what supports a model for an avidity–driven formation of the signalosome. Finally, TRAF proteins have specific affinity for CD40, and they require distinct level of oligomerization to stabilize their interaction with the receptor. These observations define a rational basis for the particular functions of TRAFs during CD40 signaling.
Activation of TRAF molecules certainly occurs via their post–translational modification, since BCR engagement alone or in combination with anti-CD40 mAb has been shown to enhance the phosphorylation of TRAF2.[31] The accepted scenario for TRAFs is that TRAF proteins remain in the cytoplasm until activation of the receptor. Then, TRAFs are recruited to the membrane, activated in the signalosome and released into the cytoplasm for propagation of the signal by interacting with numerous kinases and adaptor/regulators in various signaling pathways.[79]
3.4.2.3.3 The TRAF–CD40 interaction viewed at the three-dimensional level
Structural data from crystal of the TRAF domain of TRAF6 complexed with a peptide derived from the cytoplasmic domain of CD40 indicated that the linear sequence of CD40 bind to a surface groove on TRAF-C in an extended conformation (Figure 23).[80] The proline side–chain is fully buried into the binding pocket, orienting all other CD40 residues for optimal contact with the edge of the β-sandwich structure (Figure 23).
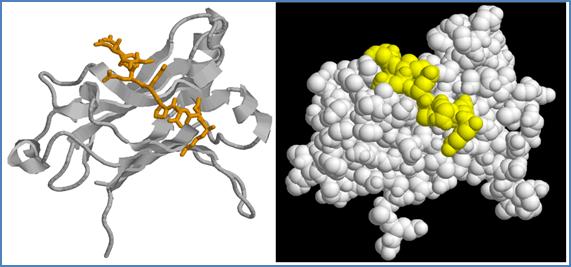
Figure 23. Three-dimensional structure of the TRAF-C domain of human TRAF6 complexed with a peptide derived from the CD40 intracytoplasmic domain. The same view of TRAF-C domain of TRAF6 complex with the CD40 fragment 230KQEPQEIDF238, that contains the Asn237-Asp mutation for an enhanced interaction affinity, represented in cartoon (left) and spacefill (right). CD40 peptide is represented in orange sticks (left) or yellow spacefill (right) (PDB code 1LB6).
TRAF2 recognition peptide bound to TRAF2 in a similar manner than TRAF6 recognition motif bound to TRAF6, apart from some variation in direction of the motif that may alter conformation of other TRAFs and thereby influence their binding to the cytoplasmic domain of CD40. Each CD40 fragment associates with one monomer of TRAF2 (Figure 24A).[81] Most importantly, ligand–induced trimerization of the receptor and self–association of TRAF molecules lead to the formation of intracellular signaling complexes with C3 symmetry with the TRAF-N domain pointing toward the cytoplasm (Figure 24B).[82] CD40L geometry is thus transduced to TRAF adaptor molecules inside the cell.
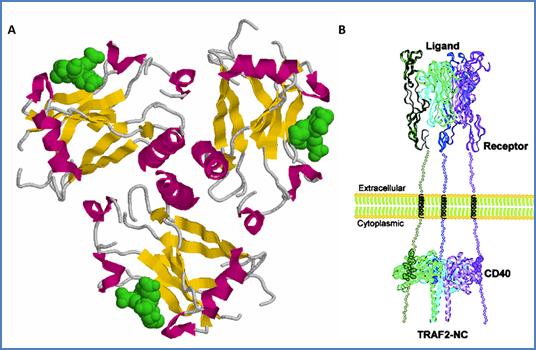
Figure 24. Three-dimensional model of the TRAF2–CD40 interaction. (A) Same structure as in Figure 22, represented in cartoons. The TRAF2–binding peptides Ac-250YPIQET255-NH2 derived from CD40 are represented in green spacefill. Three-fold symmetry axis is perpendicular to the plan, with TRAF-N domain helixes in the center pointing toward the reader. (B) Model of CD40/TRAF2 complex induced by ligand/receptor interaction as suggested by McWhirter and coworkers (PDB code 1QSC).[81]
3.4.2.4 TRAF–specific signaling
3.4.2.4.1 TRAF1
TRAF1 interacts only weakly with CD40 in the absence of TRAF2, and cooperates with TRAF2 to enhance some CD40–induced pathways.[83] It was also proposed that the stoichiometry of each protein in TRAF1–TRAF2 complexes determine their ability to engage signaling downstream CD40.[84] Although TRAF1 has been shown to associate with a number of cytoplasmic molecules implicated in NF-κB and JNK activation, its complex role in regulation of apoptotic signals is not fully understood.
3.4.2.4.2 TRAF2
TRAF2 was linked to the NF-κB and AP-1 pathways,[85] and transfection of full-length TRAF2 cells led to an enhanced activation of NF-κB in untreated epithelial cells.[63] NF-κB–inducing kinase (NIK) was shown to mediate TRAF2–dependent NF-κB activation from the LT-β receptor and to interact with TRAFs 1, 3, 5 and 6.[86] Several other molecules that influence TRAF2–mediated NF-κB activation have also been described.[87] More recently, the MAP3K8 Tpl/COT1 was shown to be recruited to the CD40 receptor complex via its interaction with TRAF2 and TRAF6, resulting in activation of inhibitor of κB (IκB) kinase (IKK) and subsequent induction of NF-κB.[88] TRAF2 can also interact with MEKK1 (also named MAP3K), and TNF-R1 activation was shown to enhance the TRAF2–MEKK1 interaction, thereby activating MAPK pathways.[89] Experiments with TRAF2-/- mice confirmed that TRAF2 is an essential mediator of JNK activation.[90]
Interestingly, TRAFs were also shown to interact with cell structure−related proteins. For example, caveolin-1 was shown to interact with TRAF2, targeting it to lipid rafts.[91]
3.4.2.4.3 TRAF3
TRAF3 overexpression was shown to block the TRAF2–dependent NF-κB induction.[63] Furthermore, a dominant negative form of TRAF3, in which the ring finger was deleted, inhibited CD40–induced upregulation of CD23 in human B lymphoma, probably by displacing TRAF2 from the receptor.[92] In fact, TRAF3 is not necessary for the induction of antibody secretion,[93] and B cells from TRAF3–deficient mice upregulated CD23 and proliferated normally in response to CD40L.[94] However, isotype switching in response to T–dependent antigens is defective, suggesting that TRAF3 is not required for CD40 signaling, but is important in T–dependent immune responses.
Recently, association between TRAF3 and TRAF2 was reported.[95] This interaction involves the TRAF-C domain of TRAF3 and zinc-finger regions of TRAF2. TRAF3/TRAF2 hetero−trimerization inhibits the TRAF2–induced NF-κB pathway, but not the activation of AP-1. TRAF3 was also shown to regulate both the classical (p50/RelA) and alternative (p52/RelB)[96] NF-κB pathways induced from TRAFs 2/5.[97] An important role for TRAF3 in CD40–mediated apoptosis of cancer cell was described recently, through activation of the JNK/AP-1 pathway and activation of caspase 9.[60]
Interestingly, recruitment of TRAF3 to microtubule network was shown to occur via MIP-T3 which specifically interacts with TRAF3 through the TRAF-N.[98] TRAF3 was also shown associated with a component of the nuclear pore central plug.[99]
3.4.2.4.4 TRAF4
TRAF4 was shown to be expressed in breast carcinoma but not in normal tissues, and localized predominantly to the nucleus,[100] and is overexpressed in human carcinomas, suggesting that TRAF4 has an oncogenic role.[101] To date, no data have described implication of TRAF4 in CD40 signaling, although a CD40–TRAF4 could be detected in vitro.[102] Nevertheless, TRAF4 mediates TNF signaling and inhibits Fas–mediated apoptosis of HEK cells when overexpressed.[103] Furthermore, TRAF4 enhances GITR–induced NF-κB activation, modulating suppressive functions of Treg cells.[104]
3.4.2.4.5 TRAF5
TRAF5 involvement in CD40 signaling was provided by cells from TRAF5-/- mice in which receptor–mediated lymphocyte activation was substantially impaired but which displayed normal CD40–mediated NF-κB and JNK activation.[105] This suggests that TRAF5 compensation mechanisms might be efficient to some extent. TRAF5 self-associates in the cytoplasm of HeLa cells,[94] and interacts with either CD40 directly[70] or via heterotrimerization with TRAF3.[67] The latter case was suggested to explain why CD40-induced NF-κB mediated by TRAF5 was enhanced in the presence of TRAF3.[106] Finally, like TRAF2, TRAF5 is important for the NF-κB activation via both the classical and alternative pathways which is regulated by TRAF3.[96]
3.4.2.4.6 TRAF6
TRAF6 is implicated in CD40–mediated IKK and NF-κB activation in monocyte/macrophage.[107] TRAF6 is also implicated in CD40 signaling in B cell and is required for the production of inflammatory cytokines through the engagement of Src/ERK1/2 pathway.[108] This is consistent with earlier observations that transfection of TRAF6 into 293 cells activated NF-κB pathway, such as with TRAF2 and TRAF5.[69] Recently, TRAF6 was shown to induce apoptosis through interaction with caspase and its activation by a RING domain-dependent mechanism.[109] Interestingly, Benson et al. showed that CD40 can protect B cells from CD95−mediated apoptosis by inhibiting caspase activation via TRAF6 and the PI3K/Akt pathway.[110]
3.4.3 TRAF–independent CD40
signaling
Janus kinase 3 (JAK3) pathway seems to be important for CD40-mediated activation of monocytes[111] and dendritic cells,[112] but not of B cells.[113] JAK3 has been shown to interact with a proline motif in the intracellular region of CD40, through two identified JAK binding–motifs (Figure 25).[114] JAK3 is phosphorylated following CD40 ligation, resulting in the phosphorylation of STAT-3, STAT-5α and STAT-6. Numerous CD40–induced pathways have been described, but some of them were neither associated to TRAF nor to JAK3 (Poster 1).
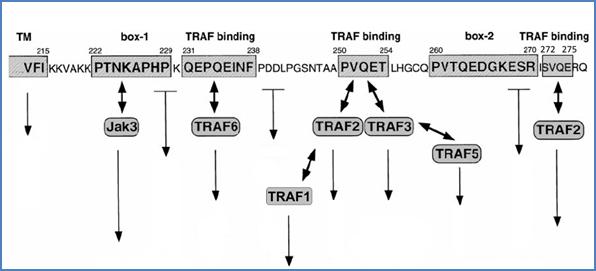
Figure 25. JAK and TRAF binding motifs on the intracytoplasmic sequence of CD40. JAK3 (box-1 and box-2) as well as TRAF (TRAF binding) binding motifs are indicated on the intracytoplasmic domain sequence of CD40. Some other pathways were shown to be initiated apart from these motifs. Figure is adapted from van Kooten et al.[7]
![]()
![]()
[1] Paulie, S, et al. (1989). The human B lymphocyte and carcinoma antigen, CDw40, is a phosphoprotein involved in growth signal transduction. J Immunol 142, 590.
[2] Grassme, H, et al. (2002). Clustering of CD40 ligand is required to form a functional contact with CD40. J Biol Chem 277, 30289.
[3] Bjorck, P, et al. (1994). Antibodies to distinct epitopes on the CD40 molecule co-operate in stimulation and can be used for the detection of soluble CD40. Immunology 83, 430.
[4] Holler, N, et al. (2003). Two adjacent trimeric Fas ligands are required for Fas signaling and formation of a death-inducing signaling complex. Mol Cell Biol 23, 1428.
[5] Fanslow, W C, et al. (1994). Structural characteristics of CD40 ligand that determine biological function. Semin Immunol 6, 267.
[6] Pound, J D, et al. (1999). Minimal cross-linking and epitope requirements for CD40-dependent suppression of apoptosis contrast with those for promotion of the cell cycle and homotypic adhesions in human B cells. Int Immunol 11, 11.
[7] van Kooten, C, et al. (2000). CD40-CD40 ligand. J Leukoc Biol 67, 2.
[8] Kaykas, A, et al. (2001). CD40 and LMP-1 both signal from lipid rafts but LMP-1 assembles a distinct, more efficient signaling complex. Embo J 20, 2641.
[9] Haswell, L E, et al. (2001). Analysis of the oligomeric requirement for signaling by CD40 using soluble multimeric forms of its ligand, CD154. Eur J Immunol 31, 3094.
[10] Pullen, S S, et al. (1999). High-affinity interactions of tumor necrosis factor receptor-associated factors (TRAFs) and CD40 require TRAF trimerization and CD40 multimerization. Biochemistry 38, 10168.
[11] Baccam, M, et al. (1999). Membrane-bound CD154, but not CD40-specific antibody, mediates NF-kappaB-independent IL-6 production in B cells. Eur J Immunol 29, 3855.
[12] Legembre, P, et al. (2003). Cutting edge: SDS-stable Fas microaggregates: an early event of Fas activation occurring with agonistic anti-Fas antibody but not with Fas ligand. J Immunol 171, 5659.
[13] Werneburg, B G, et al. (2001). Molecular characterization of CD40 signaling intermediates. J Biol Chem 276, 43334.
[14] Braesch-Andersen, S, et al. (1989). Biochemical characteristics and partial amino acid sequence of the receptor-like human B cell and carcinoma antigen CDw40. J Immunol 142, 562.
[15] Reyes-Moreno, C, et al. (2004). CD40/CD40 homodimers are required for CD40-induced phosphatidylinositol 3-kinase-dependent expression of B7.2 by human B lymphocytes. J Biol Chem 279, 7799.
[16] Challa, A, et al. (1999). Epitope-dependent synergism and antagonism between CD40 antibodies and soluble CD40 ligand for the regulation of CD23 expression and IgE synthesis in human B cells. Allergy 54, 576.
[17] Ledbetter, J A, et al. (1997). Agonistic activity of a CD40-specific single-chain Fv constructed from the variable regions of mAb G28-5. Crit Rev Immunol 17, 427.
[18] Ellmark, P, et al. (2002). Modulation of the CD40-CD40 ligand interaction using human anti-CD40 single-chain antibody fragments obtained from the n-CoDeR phage display library. Immunology 106, 456.
[19] Malmborg Hager, A C, et al. (2003). Affinity and epitope profiling of mouse anti-CD40 monoclonal antibodies. Scand J Immunol 57, 517.
[20] Ellmark, P, et al. (2003). Pre-assembly of the extracellular domains of CD40 is not necessary for rescue of mouse B cells from anti-immunoglobulin M-induced apoptosis. Immunology 108, 452.
[21] Papoff, G, et al. (1999). Identification and characterization of a ligand-independent oligomerization domain in the extracellular region of the CD95 death receptor. J Biol Chem 274, 38241.
[22] Chan, F K, et al. (2000). A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 288, 2351.
[23] Deng, G M, et al. (2005). Amelioration of inflammatory arthritis by targeting the pre-ligand assembly domain of tumor necrosis factor receptors. Nat Med 11, 1066.
[24] Siegel, R M, et al. (2000). Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science 288, 2354.
[25] Simons, K, et al. (1997). Functional rafts in cell membranes. Nature 387, 569.
[26] Hayashi, M, et al. (2006). Detection of cholesterol-rich microdomains in the inner leaflet of the plasma membrane. Biochem Biophys Res Commun 351, 713.
[27] Muppidi, J R, et al. (2004). Ligand-independent redistribution of Fas (CD95) into lipid rafts mediates clonotypic T cell death. Nat Immunol 5, 182.
[28] Hostager, B S, et al. (2000). Recruitment of CD40 and tumor necrosis factor receptor-associated factors 2 and 3 to membrane microdomains during CD40 signaling. J Biol Chem 275, 15392.
[29] Vidalain, P O, et al. (2000). CD40 signaling in human dendritic cells is initiated within membrane rafts. EMBO J 19, 3304.
[30] Malapati, S, et al. (2001). The influence of CD40 on the association of the B cell antigen receptor with lipid rafts in mature and immature cells. Eur J Immunol 31, 3789.
[31] Haxhinasto, S A, et al. (2004). Synergistic B cell activation by CD40 and the B cell antigen receptor: role of B lymphocyte antigen receptor-mediated kinase activation and tumor necrosis factor receptor-associated factor regulation. J Biol Chem 279, 2575.
[32] Pham, L V, et al. (2002). A CD40 Signalosome anchored in lipid rafts leads to constitutive activation of NF-kappaB and autonomous cell growth in B cell lymphomas. Immunity 16, 37.
[33] Bollinger, C R, et al. (2005). Ceramide-enriched membrane domains. Biochim Biophys Acta 1746, 284.
[34] Li, H, et al. (2004). Functional caveolae are a prerequisite for CD40 signaling in human renal proximal tubule cells. Am J Physiol Renal Physiol 286, F711.
[35] Grassme, H, et al. (2002). Ceramide-rich membrane rafts mediate CD40 clustering. J Immunol 168, 298.
[36] Grassme, H, et al. (2001). Molecular mechanisms of ceramide-mediated CD95 clustering. Biochem Biophys Res Commun 284, 1016.
[37] Gulbins, E, et al. (2002). Ceramide and cell death receptor clustering. Biochim Biophys Acta 1585, 139.
[38] Brenner, B, et al. (1998). Fas/CD95/Apo-I activates the acidic sphingomyelinase via caspases. Cell Death Differ 5, 29.
[39] Zeidan, Y H, et al. (2007). Activation of Acid Sphingomyelinase by Protein Kinase C{delta}-mediated Phosphorylation. J Biol Chem 282, 11549.
[40] Clark, E A, et al. (1990). Association between IL-6 and CD40 signaling. IL-6 induces phosphorylation of CD40 receptors. J Immunol 145, 1400; Inui, S, et al. (1990). Identification of the intracytoplasmic region essential for signal transduction through a B cell activation molecule, CD40. Eur J Immunol 20, 1747.
[41] Stamenkovic, I, et al. (1989). A B-lymphocyte activation molecule related to the nerve growth factor receptor and induced by cytokines in carcinomas. Embo J 8, 1403.
[42] Einfeld, D A, et al. (1988). Molecular cloning of the human B cell CD20 receptor predicts a hydrophobic protein with multiple transmembrane domains. EMBO J 7, 711.
[43] Uckun, F M, et al. (1991). Stimulation of protein tyrosine phosphorylation, phosphoinositide turnover, and multiple previously unidentified serine/threonine-specific protein kinases by the Pan-B-cell receptor CD40/Bp50 at discrete developmental stages of human B-cell ontogeny. J Biol Chem 266, 17478.
[44] Lane, P J, et al. (1991). The role of tyrosine phosphorylation in signal transduction through surface Ig in human B cells. Inhibition of tyrosine phosphorylation prevents intracellular calcium release. J Immunol 146, 715.
[45] Kansas, G S, et al. (1991). Transmembrane signals generated through MHC class II, CD19, CD20, CD39, and CD40 antigens induce LFA-1-dependent and independent adhesion in human B cells through a tyrosine kinase-dependent pathway. J Immunol 147, 4094.
[46] Knox, K A, et al. (1993). Protein tyrosine phosphorylation is mandatory for CD40-mediated rescue of germinal center B cells from apoptosis. Eur J Immunol 23, 2578.
[47] Faris, M, et al. (1994). CD40 signaling pathway: anti-CD40 monoclonal antibody induces rapid dephosphorylation and phosphorylation of tyrosine-phosphorylated proteins including protein tyrosine kinase Lyn, Fyn, and Syk and the appearance of a 28-kD tyrosine phosphorylated protein. J Exp Med 179, 1923.
[48] Lalmanach-Girard, A C, et al. (1993). T cell-dependent induction of NF-kappa B in B cells. J Exp Med 177, 1215.
[49] Berberich, I, et al. (1994). Cross-linking CD40 on B cells rapidly activates nuclear factor-kappa B. J Immunol 153, 4357.
[50] Yuan, W, et al. (1997). Programmed cell death in human ovary is a function of follicle and corpus luteum status. J Clin Endocrinol Metab 82, 3148.
[51] Inui, S, et al. (1990). Identification of the intracytoplasmic region essential for signal transduction through a B cell activation molecule, CD40. Eur J Immunol 20, 1747.
[52] Bradley, J R, et al. (2001). Tumor necrosis factor receptor-associated factors (TRAFs). Oncogene 20, 6482.
[53] Wajant, H, et al. (2001). The TNF-receptor-associated factor family: scaffold molecules for cytokine receptors, kinases and their regulators. Cell Signal 13, 389.
[54] Xu, L G, et al. (2004). TRAF7 potentiates MEKK3-induced AP1 and CHOP activation and induces apoptosis. J Biol Chem 279, 17278; Bouwmeester, T, et al. (2004). A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol 6, 97.
[55] Ardila-Osorio, H, et al. (2005). TRAF interactions with raft-like buoyant complexes, better than TRAF rates of degradation, differentiate signaling by CD40 and EBV latent membrane protein 1. Int J Cancer 113, 267.
[56] Wu, S, et al. (2005). LMP1 protein from the Epstein-Barr virus is a structural CD40 decoy in B lymphocytes for binding to TRAF3. J Biol Chem 280, 33620.
[57] Kuhne, M R, et al. (1997). Assembly and regulation of the CD40 receptor complex in human B cells. J Exp Med 186, 337.
[58] Brown, K D, et al. (2002). Regulation of TRAF2 signaling by self-induced degradation. J Biol Chem 277, 19433.
[59] Brink, R, et al. (1998). Tumor necrosis factor receptor (TNFR)-associated factor 2A (TRAF2A), a TRAF2 splice variant with an extended RING finger domain that inhibits TNFR2-mediated NF-kappaB activation. J Biol Chem 273, 4129.
[60] Georgopoulos, N T, et al. (2006). A novel mechanism of CD40-induced apoptosis of carcinoma cells involving TRAF3 and JNK/AP-1 activation. Cell Death Differ 13, 1789.
[61] Hostager, B S, et al. (2003). Tumor necrosis factor receptor-associated factor 2 (TRAF2)-deficient B lymphocytes reveal novel roles for TRAF2 in CD40 signaling. J Biol Chem 278, 45382.
[62] Arron, J R, et al. (2002). Regulation of the subcellular localization of tumor necrosis factor receptor-associated factor (TRAF)2 by TRAF1 reveals mechanisms of TRAF2 signaling. J Exp Med 196, 923.
[63] Rothe, M, et al. (1995). The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell 83, 1243.
[64] Cheng, G, et al. (1995). Involvement of CRAF1, a relative of TRAF, in CD40 signaling. Science 267, 1494.
[65] Min, W, et al. (1998). The N-terminal domains target TNF receptor-associated factor-2 to the nucleus and display transcriptional regulatory activity. J Immunol 161, 319.
[66] Wajant, H, et al. (1999). TNF receptor associated factors in cytokine signaling. Cytokine Growth Factor Rev 10, 15.
[67] Pullen, S S, et al. (1998). CD40-tumor necrosis factor receptor-associated factor (TRAF) interactions: regulation of CD40 signaling through multiple TRAF binding sites and TRAF hetero-oligomerization. Biochemistry 37, 11836.
[68] Park, Y C, et al. (1999). Structural basis for self-association and receptor recognition of human TRAF2. Nature 398, 533.
[69] Ishida, T, et al. (1996). Identification of TRAF6, a novel tumor necrosis factor receptor-associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region. J Biol Chem 271, 28745.
[70] Ishida, T K, et al. (1996). TRAF5, a novel tumor necrosis factor receptor-associated factor family protein, mediates CD40 signaling. Proc Natl Acad Sci U S A 93, 9437.
[71] Leo, E, et al. (1999). Differential requirements for tumor necrosis factor receptor-associated factor family proteins in CD40-mediated induction of NF-kappaB and Jun N-terminal kinase activation. J Biol Chem 274, 22414.
[72] Lu, L F, et al. (2003). CD40 signaling through a newly identified tumor necrosis factor receptor-associated factor 2 (TRAF2) binding site. J Biol Chem 278, 45414.
[73] Mackey, M F, et al. (2003). Distinct contributions of different CD40 TRAF binding sites to CD154-induced dendritic cell maturation and IL-12 secretion. Eur J Immunol 33, 779.
[74] Ahonen, C, et al. (2002). The CD40-TRAF6 axis controls affinity maturation and the generation of long-lived plasma cells. Nat Immunol 3, 451.
[75] Jabara, H, et al. (2002). The binding site for TRAF2 and TRAF3 but not for TRAF6 is essential for CD40-mediated immunoglobulin class switching. Immunity 17, 265.
[76] Rothe, M, et al. (1996). I-TRAF is a novel TRAF-interacting protein that regulates TRAF-mediated signal transduction. Proc Natl Acad Sci U S A 93, 8241.
[77] Chin, A I, et al. (1999). TANK potentiates tumor necrosis factor receptor-associated factor-mediated c-Jun N-terminal kinase/stress-activated protein kinase activation through the germinal center kinase pathway. Mol Cell Biol 19, 6665.
[78] Ye, H, et al. (1999). The structural basis for the recognition of diverse receptor sequences by TRAF2. Mol Cell 4, 321.
[79] Wajant, H, et al. (2001). The TNF-receptor-associated factor family: scaffold molecules for cytokine receptors, kinases and their regulators. Cell Signal 13, 389.
[80] Ye, H, et al. (2002). Distinct molecular mechanism for initiating TRAF6 signalling. Nature 418, 443.
[81] McWhirter, S M, et al. (1999). Crystallographic analysis of CD40 recognition and signaling by human TRAF2. Proc Natl Acad Sci U S A 96, 8408.
[82] Chung, J Y, et al. (2002). All TRAFs are not created equal: common and distinct molecular mechanisms of TRAF-mediated signal transduction. J Cell Sci 115, 679.
[83] Xie, P, et al. (2006). Cooperation between TNF receptor-associated factors 1 and 2 in CD40 signaling. J Immunol 176, 5388.
[84] Fotin-Mleczek, M, et al. (2004). Tumor necrosis factor receptor-associated factor (TRAF) 1 regulates CD40-induced TRAF2-mediated NF-kappaB activation. J Biol Chem 279, 677.
[85] Bradley, J R, et al. (2001). Tumor necrosis factor receptor-associated factors (TRAFs). Oncogene 20, 6482.
[86] Luftig, M A, et al. (2001). Effects of the NIK aly mutation on NF-kappaB activation by the Epstein-Barr virus latent infection membrane protein, lymphotoxin beta receptor, and CD40. J Biol Chem 276, 14602.
[87] Song, H Y, et al. (1996). The tumor necrosis factor-inducible zinc finger protein A20 interacts with TRAF1/TRAF2 and inhibits NF-kappaB activation. Proc Natl Acad Sci U S A 93, 6721.
[88] Chan, H, et al. (2005). TRAF-dependent association of protein kinase Tpl2/COT1 (MAP3K8) with CD40. Biochem Biophys Res Commun 328, 198.
[89] Baud, V, et al. (1999). Signaling by proinflammatory cytokines: oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev 13, 1297.
[90] Yeh, W C, et al. (1997). Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity 7, 715.
[91] Feng, X, et al. (2001). Caveolin-1 associates with TRAF2 to form a complex that is recruited to tumor necrosis factor receptors. J Biol Chem 276, 8341.
[92] Cheng, G, et al. (1995). Involvement of CRAF1, a relative of TRAF, in CD40 signaling. Science 267, 1494.
[93] Hostager, B S, et al. (1999). Cutting edge: contrasting roles of TNF receptor-associated factor 2 (TRAF2) and TRAF3 in CD40-activated B lymphocyte differentiation. J Immunol 162, 6307.
[94] Xu, Y, et al. (1996). Targeted disruption of TRAF3 leads to postnatal lethality and defective T-dependent immune responses. Immunity 5, 407.
[95] He, L, et al. (2004). TRAF3 forms heterotrimers with TRAF2 and modulates its ability to mediate NF-{kappa}B activation. J Biol Chem 279, 55855.
[96] Dejardin, E. (2006). The alternative NF-kappaB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem Pharmacol 72, 1161.
[97] Hauer, J, et al. (2005). TNF receptor (TNFR)-associated factor (TRAF) 3 serves as an inhibitor of TRAF2/5-mediated activation of the noncanonical NF-kappaB pathway by TRAF-binding TNFRs. Proc Natl Acad Sci U S A 102, 2874.
[98] Ling, L, et al. (2000). MIP-T3, a novel protein linking tumor necrosis factor receptor-associated factor 3 to the microtubule network. J Biol Chem 275, 23852.
[99] Gamper, C, et al. (2000). TRAF-3 interacts with p62 nucleoporin, a component of the nuclear pore central plug that binds classical NLS-containing import complexes. Mol Immunol 37, 73.
[100] Regnier, C H, et al. (1995). Presence of a new conserved domain in CART1, a novel member of the tumor necrosis factor receptor-associated protein family, which is expressed in breast carcinoma. J Biol Chem 270, 25715.
[101] Camilleri-Broet, S, et al. (2007). TRAF4 overexpression is a common characteristic of human carcinomas. Oncogene 26, 142.
[102] Roy, N, et al. (1997). The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. Embo J 16, 6914.
[103] Fleckenstein, D S, et al. (2003). Tumor necrosis factor receptor-associated factor (TRAF) 4 is a new binding partner for the p70S6 serine/threonine kinase. Leuk Res 27, 687.
[104] Esparza, E M, et al. (2004). TRAF4 functions as an intermediate of GITR-induced NF-kappaB activation. Cell Mol Life Sci 61, 3087.
[105] Nakano, H, et al. (1999). Targeted disruption of Traf5 gene causes defects in CD40- and CD27-mediated lymphocyte activation. Proc Natl Acad Sci U S A 96, 9803.
[106] Leo, E, et al. (1999). Differential requirements for tumor necrosis factor receptor-associated factor family proteins in CD40-mediated induction of NF-kappaB and Jun N-terminal kinase activation. J Biol Chem 274, 22414.
[107] Mukundan, L, et al. (2005). TNF receptor-associated factor 6 is an essential mediator of CD40-activated proinflammatory pathways in monocytes and macrophages. J Immunol 174, 1081.
[108] Lomaga, M A, et al. (1999). TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev 13, 1015.
[109] He, L, et al. (2006). TRAF6 regulates cell fate decisions by inducing caspase 8-dependent apoptosis and the activation of NF-kappaB. J Biol Chem 281, 11235.
[110] Benson, R J, et al. (2006). Rapid CD40-mediated rescue from CD95-induced apoptosis requires TNFR-associated factor-6 and PI3K. Eur J Immunol 36, 2535.
[111] Revy, P, et al. (1999). Activation of the Janus kinase 3-STAT5a pathway after CD40 triggering of human monocytes but not of resting B cells. J Immunol 163, 787.
[112] Saemann, M D, et al. (2002). CD40 triggered human monocyte-derived dendritic cells convert to tolerogenic dendritic cells when JAK3 activity is inhibited. Transplant Proc 34, 1407.
[113] Jabara, H H, et al. (1998). Role of JAK3 in CD40-mediated signaling. Blood 92, 2435.
[114] Hanissian, S H, et al. (1997). Jak3 is associated with CD40 and is critical for CD40 induction of gene expression in B cells. Immunity 6, 379.